现行的微生态研究最常用的两把利器非扩增子测序和宏基因组测序莫属,扩增子测序便宜,可以进行物种分类、物种丰度、揭示环境因子和微生物群落之间的相互关系,但是不能进行基因预测及功能注释。宏基因组测序不但能够获得环境物种组成和丰度信息,还能进行基因预测及功能注释,比较样品间的基因丰度差异和代谢网络差异,但是宏基因组测序价格贵,目前最常用的策略就是同时采用扩增子测序和宏基因组测序从而综合得到两种技术的优势结果。下面我们就总结一下这种研究策略及它的具体应用。
策略:扩增子测序+宏基因组测序
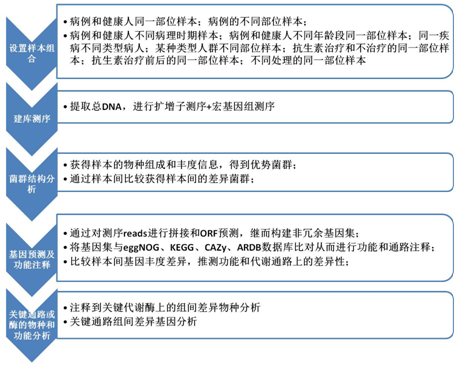
扩增子测序+宏基因组测序案例1:中样本量
英文题目:Diet shapes the gut microbiome of pigs during nursing and weaning
中文题目:哺乳期及断奶期不同饮食分别对小猪肠道菌群的影响
期刊名:Microbiome 发表时间: 2015 IF:9
一、材料方法
研究材料:选择3头母猪刚出生的27头小猪,先母乳喂养21天,在小猪出生后的第1、3、5、7、14、21、28、35和42天用无菌棉拭子收集其粪便样品,共243个样本,进行16S扩增子测序 ;出生21天后开始用饲料喂养小猪,每窝选择1头小猪,共3头小猪,分别收集其断奶前(出生后14日)和断奶后(出生后35日)的粪便样品,分别对这6个样本进行宏基因组测序。
技术方法:16S扩增子:Illumina Miseq测序平台(PE250),V4区;宏基因组:Illumina Miseq测序平台(PE150)
分析内容:α多样性分析、Beta多样性分析、群落结构分析、组间功能和通路差异分析、注释到关键代谢酶上的物种分布
二、研究结果:
1.α多样性分析:质量控制后,16S 扩增子测序共产生2966033 个reads,平均每个样本产生13067个reads,α多样性分析表明断奶后饲料喂养的小猪肠道微生物多样性明显比断奶前要高(图1b),从出生到断奶,微生物多样性慢慢增加,到断奶的第21天达到平台期(图1a)。
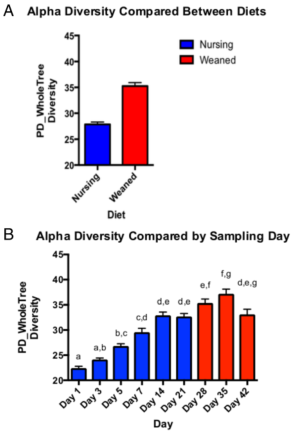
图1:不同喂养方式的小猪微生物PD分析(a)和不同喂养天数的小猪微生物PD分析(b)
2. Beta多样性分析:PCA分析表明母乳喂养和饲料喂养的小猪肠道菌群结构能明显分开,说明两者的差异较大(图2a);但是出生后不同天数小猪的肠道菌群结构则不能明显分开(图2b-c)。
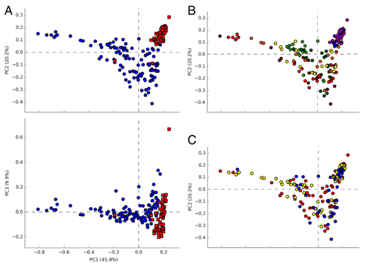
图2.不同喂养方式的小猪微生物PCA分析(a)和不同喂养天数的小猪微生物PCA分析(b)(图a蓝色代表母乳喂养样品,红色代表饲料喂养样品)。
3. 群落结构分析:令人惊讶的是,母乳喂养过程中1-21天的微生物群落结构是相对稳定的,主要富集在拟杆菌科、梭菌科、毛螺菌科等;断奶后饲料喂养相对于母乳喂养普雷沃氏菌科增加了50倍,瘤胃菌科和乳杆菌科等显著增加;而类杆菌科显著降低(图3)。同时也找到了母乳喂养过程和饲料喂养过程小猪肠道微生物群落中的主要差异物种(图4)。
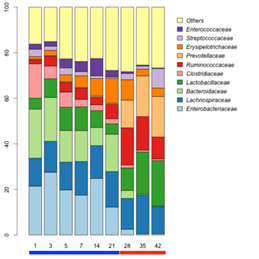
图3. 不同时间点样品群落结构组成分析
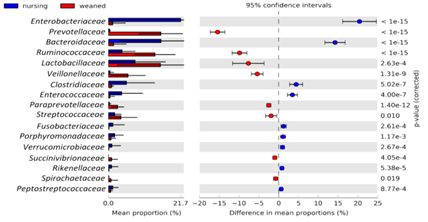
图4.两组样本间物种差异比较分析
4. 组间功能和通路差异分析:
宏基因组测序平均每个样本得到497万个reads,质量控制和过滤掉宿主基因组后,进行物种注释,PCA分析能够把母乳喂养过程和饲料喂养过程小猪肠道样本明显区分开(图a)。通过功能注释发现,母乳喂养过程和饲料喂养过程小猪肠道菌群在糖代谢方面有显著差异。母乳喂养的小猪肠道菌群主要富集在乳糖代谢、N-乙酰氨基葡萄糖代谢、半乳糖代谢、唾液酸代谢等过程(图b)。饲料喂养小猪肠道菌群主要富集淀粉,β-葡聚糖木糖和阿拉伯糖降解酶等过程(图c)。
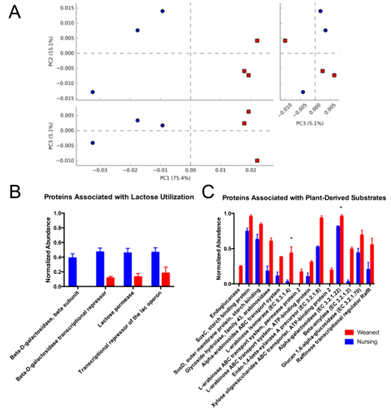
图5.宏基因组测序的6个小猪肠道样本的PCA分析(a),和参与乳糖代谢(b)和植物多糖代谢(c)的基因丰度比较。
5. 注释到关键代谢酶上的物种分布:
关键代谢步骤(饮食中糖的代谢)的不同被认为与饮食相关的不同菌群结构的主要原因。例如母乳喂养过程,降解乳糖的主要是唾液酸酶和β-己糖胺酶,参与此过程的以类杆菌属为主。在饲料喂养过程,参与植物多糖降解的以β-木糖苷酶、内-1,4-β-木聚糖酶和α-N-阿拉伯呋喃糖酶为主,参与此过程的菌种更加广泛,单仍以类杆菌属为主。
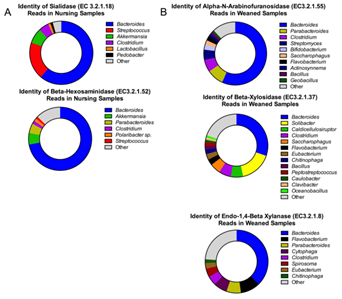
图6. 断奶前(a)或断奶后(b)猪肠道样本注释到关键代谢酶上的宏基因组序列物种丰度分布
扩增子测序+宏基因组测序案例2:小样本量
英文题目:Tongue Coating and the Salivary Microbial Communities Vary in Children with Halitosis
中文题目:舌苔,唾液微生物群落变化与儿童口臭
期刊名:Scientific Reports 发表时间: 2016 IF:5.228
一、材料方法
研究材料:10个4-5岁健康儿童的舌苔(TH);10个4-5岁健康儿童的唾液(SH);10个4-5岁口臭儿童的舌苔(TD);10个4-5岁口臭儿童的唾液(TH)。40个样本分别进行16S扩增子测序;4组样本分别混池,然后对这4个混池样本进行宏基因组测序。
技术方法:16S扩增子:454 GS FLX Titanium测序平台,V1-V3区;宏基因组:Illumina HiSeq 2500测序平台(PE150)
分析内容:α多样性分析、群落结构分析、Beta多样性分析、组间功能和通路差异分析、某通路组间差异分析
二、研究结果:
1.α多样性分析:质量控制后,16S 扩增子测序共产生394,697个reads,平均每个样本产生10,387 ± 1,907个reads,平均每个样本得到66–266 个OTUs。α多样性分析表明口臭儿童舌苔样本的物种多样性明显高于健康儿童的舌苔样本(P<0.05,图 1A),但口臭儿童和健康儿童的唾液样本之间物种多样性则没有显著差异(P>0.05,图 1B)。
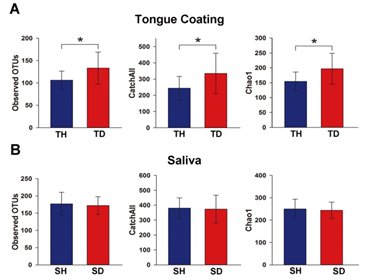
图1.健康儿童和口臭儿童α多样性指数比较分析,健康儿童的舌苔:TH;健康儿童的唾液:SH;口臭儿童的舌苔:TD;口臭儿童的唾液:TH
2. 群落结构分析:8个OTUs相对丰度在口臭儿童舌苔样本中显著增高(图 2A):其中Leptotrichia wadei (OTU398) 和Peptostreptococcus stomatis (OTU135) 在口臭儿童舌苔样本中显著增高并且在所有口臭儿童舌苔样本中都检测到(图 2B)。9个OTU的丰度在健康儿童和口臭儿童的唾液样本中有显着差异(图 2C):其中4个OTUs (L. wadei (OTU398),Prevotella shahii (OTU283),TM7 genus incertae sedis(OTU199) ,Solobacterium moorei (OTU30)) 在口臭儿童唾液样本中显著增高,但是只有P. shahii(otu283)多见于口臭儿童的唾液样本(图 2D)。
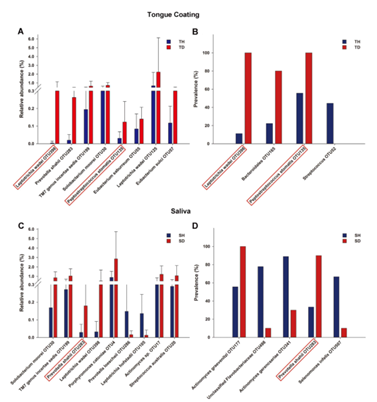
图2.不同OTUs在健康和口臭样品中之间的相对丰度和患病率差异
3. Beta多样性分析:PCA分析发现一个趋势:无论在唾液还是舌苔样本中,健康儿童的样本聚集的更紧密,而口臭儿童的样本更分散。并且有两个健康儿童样本(蓝色方块4和5)落到口臭儿童样本中,预示它有患口臭的风险。
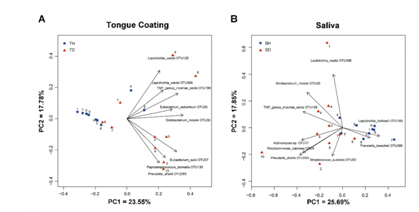
图3.健康和口臭样品的PCA分析
通过皮尔森相关系数获得很多有相关性的OTUs,然后绘制健康儿童和口臭儿童唾液、舌苔微生物的共线网络图,25个唾液节点和2个舌苔节点在健康组间被筛选到(图 4A)。23个舌苔节点和4个唾液节点在口臭组间被筛选到(图4B)。同样的而结果存在于组内(图4C,D),这些结果在健康和口臭的条件下唾液和舌苔中的微生物菌群存在不同的互作模式:健康人中起主要作用的是唾液中的OTUs,口气儿童中起主要作用的是舌苔中的OTUs。
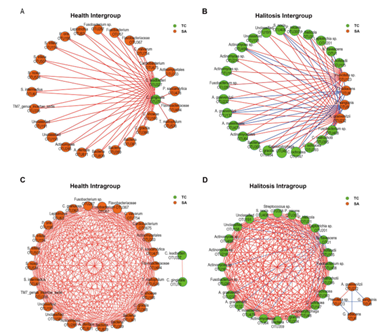
图4.在健康和口臭的条件下唾液和舌苔中的微生物菌群存在不同的互作模式。绿色代表舌苔样品中OTUs,红色的点代表唾液样品中OTUs。这些相关性分为组间相关性(即舌苔和唾液之间OTUs的相关性)(A,B),组内相关性(即舌苔样本内OTUs的相关性或唾液样本内OTUs的相关性)(C,D)。
4. 组间功能和通路差异分析:4个混池样本宏基因组测序共得到30.3 Gb数据,通过去除宿主基因组,基因拼接和基因预测,发现很多基因被口臭和健康儿童所共享。通过差异倍数>5和DEGexp(P<0.001)两种方法取交集筛选到口臭儿童和健康儿童中的差异基因(图5A,B),接着对这些差异基因进行KEGG/eggNOG注释,结果发现“传染病:细菌”和“酮类化合物与萜类化合物代谢”这两种通路的基因在口臭儿童中显著富集。
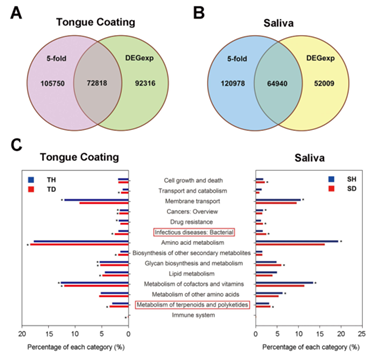
图5 口臭和健康儿童样品中功能和通路差异分析
5. 某通路组间差异分析:细菌产生H2S有两条途径:硫酸盐降解以及半胱氨酸和蛋氨酸的desulphydration,通过比较口臭儿童和健康儿童中的差异基因,发现调控硫酸盐降解的cysC在口臭儿童样本中丰度更高(图 6)。另外促进半胱氨酸和蛋氨酸desulphydration过程的cbs、aspB、yhdR、sseA 、glpE在口臭儿童样本中基因丰度更高(图 6),而编码半胱氨酸合酶B,降解H2S 转化成半胱胺酸的cysM在在口臭儿童样本中基因丰度较低,以上两种结果造成在口臭儿童样本中相对于健康人中产生更高水平的H2S。
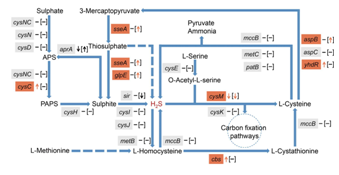
图6 口臭和健康儿童样品中H2S代谢通路差异分析。差异倍数>2定为差异基因,用箭头表示。向上的箭头表示基因在口臭样品中更丰富,向下的箭头表示基因在口臭样本比较少。差异倍数< 2的基因用“−”表示。舌苔样本中的变化用在“[ ]”外面表示,唾液样本中的变化用在“[ ]”内部表示。