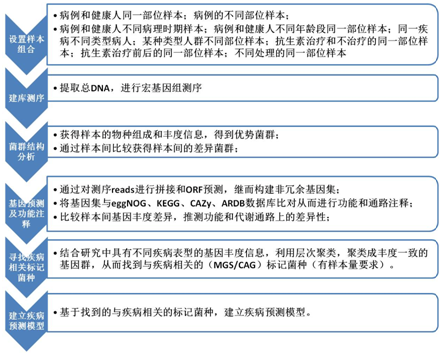
目前的微生态研究分为微生态与健康、微生态与环境、微生态与农牧三个方向。从大型微生物组的研究领域来看,人类微生物组所占比例最大, 按照微生物群在人微生态系统中的空间不同可以把人体微生态系统分为以下几类:胃肠道微生态系统、口腔微生态系统、生殖道微生态系统、皮肤微生态系统、呼吸道微生态系统。近年来,宏基因组测序为人体微生态研究提供新的方向和内容,下面我们分别根据样本量大小总结了目前最经典也是最广泛使用的组间宏基因组测序研究策略。
研究策略:组间宏基因组学
组间宏基因组学案例1:大样本量
英文题目:Alterations of human gut microbiome in liver cirrhosis
中文题目:肝硬化中肠道菌群的改变
期刊名:Nature 发表时间: 2014 IF:38.138
一、材料方法
研究材料:收集了中国汉族123例肝硬化患者和114个健康人粪便样本,其中实验阶段98 个肝硬化患者及 83 个健康人的粪便样本;验证阶段收集了 31个健康人和25个肝硬化患者的粪便样本
技术方法:Illumina HiSeq2000测序平台(PE100),平均每个样本4.75GB数据量
分析内容:数据质控、序列拼接组装、基因集构建、群落结构组成差异分析、基因丰度分析、MGS分析、组间功能和通路差异分析、疾病诊断模型建立
二、研究结果:
1.肠道菌群基因集构建
通过序列组装,基因预测,建立了肝硬化肠道菌群基因集,包含269万个非冗余基因,其中36.1%为首次发现的基因。和欧洲人(MetaHIT)、美国人(HMP)、中国糖尿病(T2D)三个基因集进行了比较,结果发现有674131个基因是四个基因集所共有的,而79万个基因是肝病基因集所特有的。
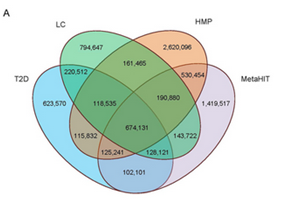
图1. 四个人类主要微生物基因集的韦恩图
2.肝硬化病人与健康人的菌落结构差异分析
在不同分类水平上(门、属、种)的对肝硬化病人与健康人的物种丰度差异进行分析,例如在门水平上,肝硬化病人和健康人中,拟杆菌门和厚壁菌门均为优势菌,相比于健康人,肝硬化病人中的拟杆菌门丰度低(图2a),但是变形菌门和梭菌门丰度高(图2b)。
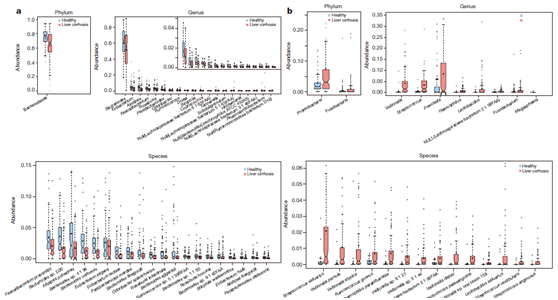
图2. 肝硬化病人和健康人在不同分类水平上的物种丰度差异。相对于健康人,在门、属、种水平上肝硬化病人丰度减少的物种(a)和丰度增加的物种(b)
3.发现和肝硬化密切相关的细菌:
根据基因丰度差异,找到肝硬化病人和健康人间显著差异的基因,利用层次聚类,聚类成丰度一致的基因群,从而找到与肝硬化相关的(MGS)标记菌种,得到了66个MGS(物种),其中38个细菌在健康人中富集,28 个细菌在肝硬化病人中富集。

图3. 分别在肝硬化病人和健康人中富集的MGS
4. 肝硬化病人和健康人功能和通路差异:eggNOG功能注释发现,相对于健康人,肝硬化病人在翻译,核糖体结构和合成等功能富集的基因更多;KEGG通路注释发现,相对于健康人,肝硬化病人在跨膜运输通路富集的基因更多。
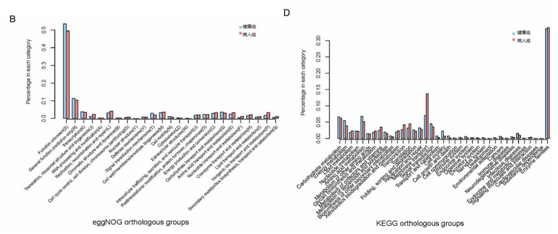
图4. 肝硬化病人和健康人功能和信号通路差异
5.建立识别肝硬化病人的的诊断模型:基于mRMR 方法到15 个高特异性和灵敏性的微生物基因标记物,提出了诊断肝硬化疾病的模型(PDI)(图a);实验阶段和验证阶段的AUC值分别为0.918和0.838,表明此模型很准确(图b.c);接着在独立验证样本中对PDI辨别肝硬化的能力进一步验证,结果发现能够利用此模型有效鉴定肝硬化患者(图d)。
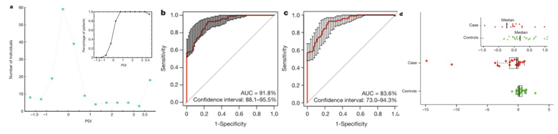
图5. 基于标记基因物建立疾病诊断模型,该模型能有效辨别肝硬化病人和健康人
比较宏基因组案例2:小样本量
英文题目:In silico analyses of metagenomes from human atherosclerotic plaque samples
中文题目:人动脉粥样硬化斑块样本的宏基因组学分析
期刊名: Microbiome 发表时间: 2015 IF:9
一、材料方法
研究材料:(1)有症状的不稳定动脉粥样硬化斑块(case):是从15例由于近期经受短暂脑缺血或轻微中风患者的颈动脉内膜切除术中获得的。(2)无症状的稳定动脉粥样硬化斑块(control):是从7个患者的尸检中获得,这组患者的死因与心血管疾病无关。取3–5毫米厚的组织,立即储存于−80 °C。
技术方法:: Illumina HiSeq2000测序平台(PE100)
分析内容:测序数据处理、Alpha多样性指数分析、Beta多样性分析、群落结构组成差异分析、组间功能和通路差异分析
二、研究结果:
1. 测序数据处理:所有22个动脉斑块样品共产生2610268774条reads,然后把这些序列和人类Hg19基因组比对,没有比对上Hg19基因组的reads平均数是884727044(每个样本平均有33.89%)。然后将这些non-hg19 reads与NCBI非冗余蛋白数据库比对。
2. Alpha多样性指数分析:从多样性指数分析(表1),稀释性曲线(图1)明显可以看出22个样本中有2个样本(样本233 和 238)比其它样本有更多的物种数。
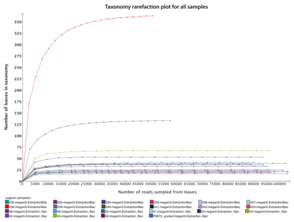
图1:22个样品(不稳定斑块样品和不稳定斑块样品)的物种稀释性曲线
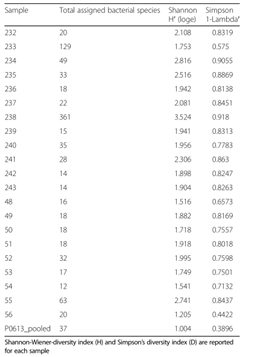
表1:多样性指数分析
3. Beta多样性分析: PCoA分析和UPGMA树展现不稳定斑块样品和不稳定斑块样品分别聚类在一起(图2)。


图2:22个样品(不稳定斑块样品和不稳定斑块样品)的PCoA分析图和UPGMA树。不稳定的动脉粥样硬化斑块样品用白色圆圈表示,7个稳定斑块样品(对照组)用灰色圆圈表示。
4. case/control群落结构组成差异分析:图3描述了所有22个样品属水平的层次聚类结果。相比于不稳定动脉粥样硬化斑块,稳定硬化斑块有更高的宿主微生物丰度,例如紫单胞菌科、链球菌科、拟杆菌科、微球菌科。相反的是,相对于稳定斑块样品,不稳定斑块样品有更多病原微生物,如螺旋杆菌科、奈瑟氏球菌科、硫发菌科。图4展示了所有样本中丰度最高的25个菌种,不稳定斑块样品中丰度最高的是鼠李糖乳杆菌MTCC 5462,多糖奈瑟球菌ATCC 43768,幽门螺旋杆菌Hp P-4,并且这些菌的丰度在不稳定斑块样品和不稳定斑块样品中存在差异,提示这些菌可能参与促进无症状动脉粥样硬化斑块向有症状动脉粥样硬化斑块的转变。
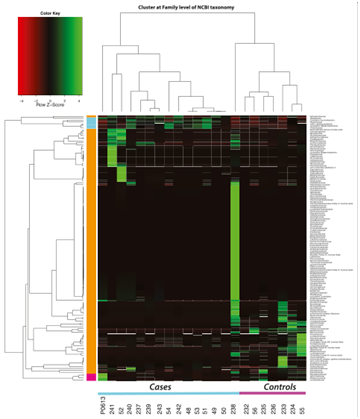
图3:来自15例有症状动脉粥样硬化性患者(实验组)的不稳定动脉粥样硬化斑块和7例死于其它原因患者(对照组)的稳定斑块的分层聚类结果。
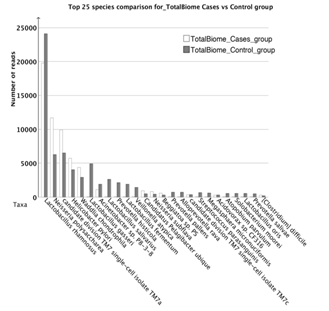
图4:所有样本中丰度最高的25个菌种,不稳定的动脉粥样硬化斑块样品用白色表示,稳定斑块样品用灰色表示。
5.case/control的功能和通路差异分析:使用SEED和KEGG对生物群落进行功能分析,结果发现在基础代谢和疾病通路方面不稳定斑块样品和不稳定斑块样品之间存在明显差异(图5),并且都参与“碳水化合物”、“氨基酸”和“能量”等有关代谢。另外也参与“心血管疾病“、“循环系统”、“免疫系统”、“细胞生长与死亡”与“传染病”等疾病相关通路。
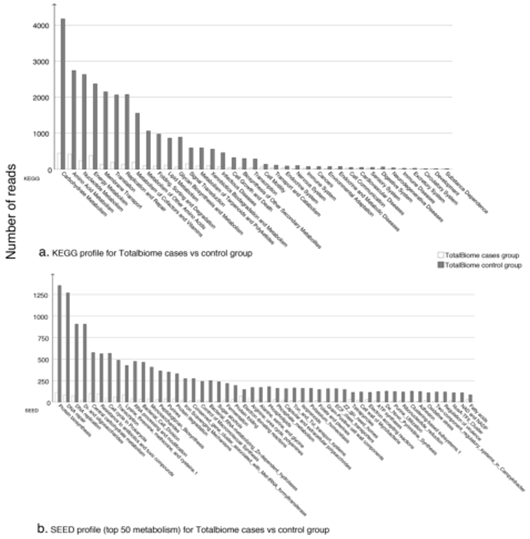
图5:使用SEED(a)和KEGG(b)分类对生物群落进行功能分析,不稳定的动脉粥样硬化斑块样品用白色表示,稳定斑块样品用灰色表示。