进行性肌无力和运动障碍
进行性肌无力和运动障碍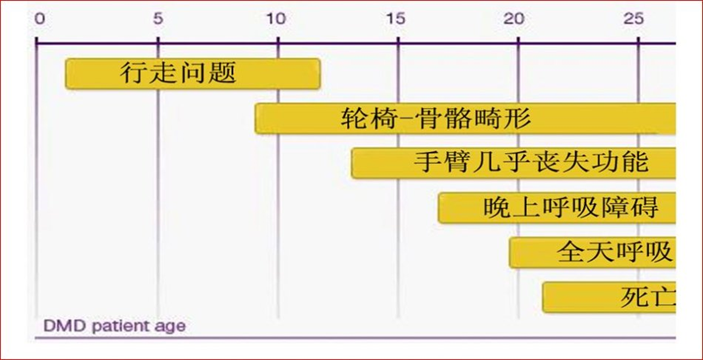 假肥大和广泛性肌萎缩
假肥大和广泛性肌萎缩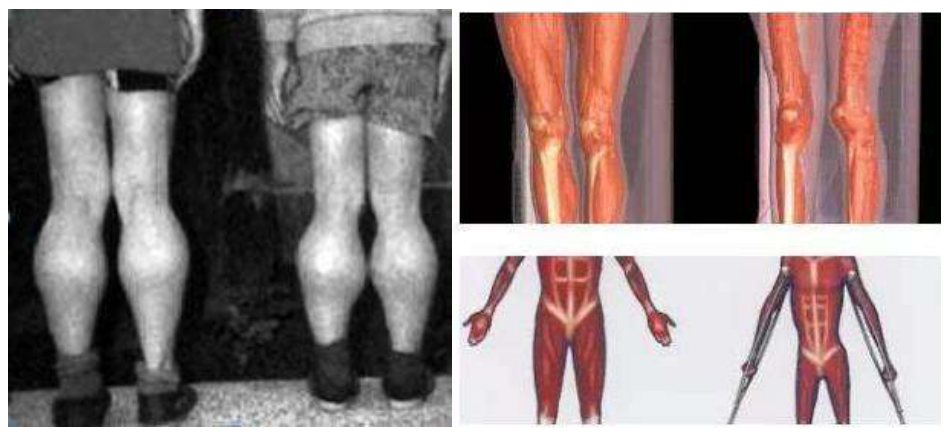 Gower征
Gower征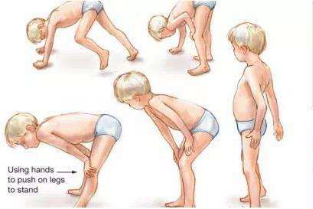
 实验室检查
实验室检查
(1) 血清磷酸肌酸激酶(CK)显著增高。
肌酶谱检查:DMD患者血液中的肌酸激酶(Creatine Kinase,CK)相当高,通常会高出正常值的数十倍甚至上百倍。骨骼肌的破坏也引起测试指标中谷丙转氨酶 (ALT)、谷草转氨酶(AST)及血清乳酸脱氢酶(LDH)的升高数倍以上。
(2) 肌电图呈典型肌病表现,周围神经传导正常。
(3) 肌肉活检,可见肌源性损害的表现。
确诊
遗传学检查(基因检测)或对活检肌肉组织进行抗肌萎缩蛋白的组织免疫化学诊断。鉴于肌肉活检是一种有创伤性的检查,基因检测对于肌肉活检有着不可替代的优势。