就在天昊生物自主研发的基于illumina高通量测序平台的--SSRseqTM产品线上市近两年,多项科研服务项目顺利完成之际,德国和奥地利研究人员的类似方法报道才“姗姗来迟”,成功发表在10月25日Ecology and Evolution杂志上。这不仅从侧面反映了天昊生物SSRseqTM技术的可行性与前瞻性,也再次印证了我们敢为人先、坚持创新驱动发展的一贯理念。下面就让我们来仔细看一下这篇文章的研究内容。

本研究提出了一个SSR-seq的名词(我们的命名为SSRseqTM),认为与传统的仅仅依靠片段大小来进行评分和基因分型的方法相比,利用下一代测序技术进行SSR基因分型,可以获得更高水平基因多态性结果。
摘要速览
简单重复序列(SSR,或称微卫星、STR)是群体遗传学中广泛使用的一种分子标记。目前,对其基因分型的检测仍然主要依靠片段长度进行。随着下一代测序(NGS)的发展,研究者获得感兴趣基因座的序列信息变得更加容易,依靠测序数据进行SSR基因型检测,避免因为片段大小近似而产生的错误判读。本研究描述了一种NGS研究策略,该策略可以对感兴趣的SSR位点进行定制化检测,一次性完成对数百个个体的基因型分析,将多重PCR、条形码和illumina测序相结合。本研究创建了三个不同的数据集,根据(a)重复区域的长度、(b)总片段长度和(c)序列一致性对等位基因进行解析,用来评估与传统检测方法的差别,计算遗传多样性及FST和RST值。通过下一代测序获得的信息提供了比传统SSR基因分型更好的分辨率,并允许更精确的进化解释。
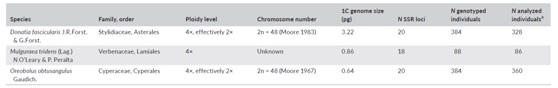
本研究利用三种被子植物:Donatia fascicularis (柱花草科)、Mulguraea tridens (马鞭草科)和Oreobolus obtusangulus (莎草科),总共对859个个体的58个SSR位点进行了基因型检测及统计分析。
表1、研究物种列表
利用illumina MiSeq2000平台paired-end模式测序。预定义的重复单位长度为3-6个碱基,微卫星区域的最小长度为21 bp。在设计引物时,其中一个引物总是被选择靠近SSR,以确保在SSR分析期间,单个read包含整个重复区域,从而确保配对reads的正确合并,目标长度可达450 bp。
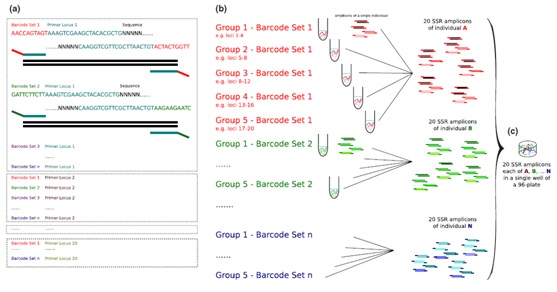
图1、实验流程图
(a)引物条形码:物种A的n×96个个体在20个基因座上进行基因分型,n:条形码引物组数;(b)多组别多重PCR;(c) SSR扩增子混合建库
表2、每个位点片段大小的非同源相似性
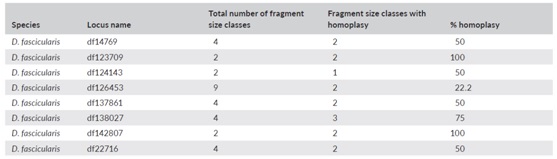
备注:非同源相似性(Homoplasy)定义为包含未检出的隐藏变异的片段大小(即长度相同但序列不同的等位基因)的数量除以片段大小的总数
序列数据显示片段大小的非同源相似性现象非常普遍(表2)。物种之间的差异(所有基因座的平均值为44.7% - 63.5%),以及一个物种内的基因座之间的差异从20%到100%不等,这是因为片段大小类别的数量与序列变异性与大小类别总数的比率不同。对于侧翼区域,SNP变异比Indel变异更普遍,许多SSR基因座也包含突变的SSR基序(表3)。
表3、位点检测信息
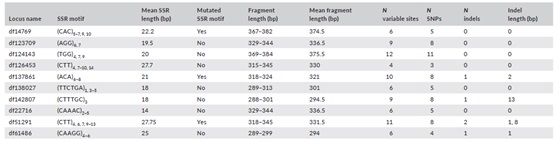
备注:SSR motif为SSR基序的序列,数字表示基序重复的次数。Variable sites包括SSR区域本身和侧翼区域的序列变异加上重复基序数量的变异。N分别表示Variable sites、SNPs和indes的数量
利用SSR-seq方法,研究者还可以统计了等位基因总数和稀有等位基因数目(表4)。
表4、基于SSR长度、片段长度(Fr length)和序列一致性(Seq identity)的三个不同数据集的等位基因总数和稀有等位基因数目(在各自的数据集中仅出现一次)
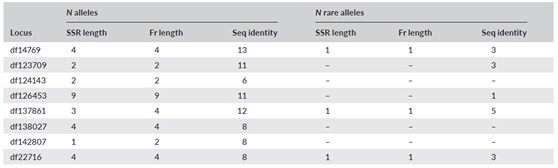
表5、基于SSR长度、片段长度(Fr length)和序列一致性(Seq identity)的三个不同数据集的遗传多样性指数
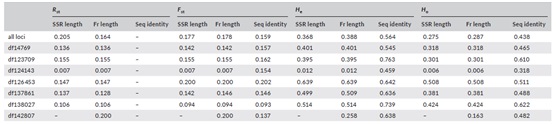
备注:He是根据样本量修正的基因多样性。Ho是观察杂合度。
通过表4和表5看出,三种数据集等位基因NA、He和Ho不尽相同。虽然SSR长度和片段长度数据集的结果较为相似,但在所有三种物种中,序列一致性数据集的遗传多样性估计值普遍较高。大多数位点FST的结果较为相近,但是也有部分位点也存在显著差异。而平均置换RST值也在不同物种间存在差异。
可以看出,序列一致性数据集在等位基因上的更高分辨率显而易见,其本身在基因座之间也是高度可变的。也就是说,三个物种通过序列数据集计算的等位基因数量分别是从SSR长度数据集计算的等位基因数量的1.2 - 8.0倍、1.3 - 13倍和1.6 - 4.0倍。
本研究最终结论认为,与传统的检测方法相比,增加的序列信息有助于更好地理解SSR变异过程。本研究结果很好的证明了,与传统的仅仅依靠片段大小来进行评分和基因分型的方法相比,利用下一代测序技术进行SSR基因分型,可以获得更高水平基因多态性结果。SSR基因座测序是一种前瞻性方法,能够检测物种内和物种间的变异,适用于大规模和精细规模系统地理学研究。
关于天昊:
天昊生物长期从事基因及遗传分析,可以提供包括SSR检测在内的多项基因检测服务。天昊生物自主研发的基于二代测序技术的SSR检测新方法--SSRseqTM,这种方法几乎克服了现存所有电泳检测方法的不足,尤其适合对多SSR位点、超高深度的分型,准确度高,并且分辨率达到单碱基的水平。因此适合所有二倍体人类、动植物、真核微生物,以及多倍体物种的SSR基因型分析。欢迎联系我们具体咨询!邮箱:techsupport@geneskies.com 电话:400-065-6886