正如4G到5G,吸引我们的不仅仅是下载网速几十倍、上百倍的提升,更是随之而来的远程会议、移动医疗、智能家居、工业控制、车联网、环境监测等一系列“万物互联”的前景应用。同样的,单细胞测序带给我们的,也绝不单单是对每个细胞层面遗传信息的简单检测与记录,而是从一个全新视角,更精准的诠释机体组成单元活动的各种进程及异质性问题,是一种创新性的理念与方法。
正因如此,近几年关于单细胞测序的研究成为了绝对的热点之一。特别是10x Genomics单细胞平台的商用化,从技术平台上实现了单细胞转录组测序(scRNA-seq)等与生物研究内容间的高效对接,在鉴定细胞聚类及类型,确定不同细胞中高表达基因及功能,深入筛选和挖掘细胞标志物,鉴定时空进程中细胞的遗传变化情况,以及利用拟时轨迹推断细胞发育分化过程等方面,都有着广泛的应用。
今天则跟大家分享刚刚在Genome Biology发表的一篇文章,通过对单细胞转录组测序数据的分析,揭示了基因调控网络间的可塑性问题,拓展了单细胞转录组测序的分析内容。
Single-cell transcriptomics unveils gene regulatory network plasticity
题目:单细胞转录组揭示了基因调控网络的可塑性
发表杂志:Genome Biology 影响因子:13.214 发表日期:2019-6

研究背景:
scRNA-seq是探索组织异质性、揭示分化动态和量化转录随机性的最新技术。目前,scRNA-seq侧重于对特征性描述的改进,如聚类、检索标记基因和探索分化轨迹。这些都基于一个分割的、区隔化的原则,即其中每个单元都是独立的。最近的大规模细胞图谱通常会到达数百个分层(子)聚类。这无疑提高了人们对各种生物环境中细胞多样性的理解。而本研究则从统一而非分割的角度出发,通过对scRNA-seq数据的挖掘,获得了新的基因网络调控信息,拓展了对单细胞数据的深入认识。
我们知道,传统的转录组分析方法(Bulk RNA-seq),可以用于推断和表征基因的调控网络,但是单细胞数据受到一系列技术限制的影响,例如缺失事件(drop-out event,即表达的基因未被scRNA-seq检测到)和高水平的噪音问题,使得利用这类数据很难进行调控网络的推断。
本文中,研究者利用scRNA-seq数据集,对调控网络分析的可行性和价值进行了探讨,提出了一种新的相关性度量方法,用来检测基因与基因之间的相关性,并且成功应用这一新方法对11个小鼠器官,健康个体和2型糖尿病患者的胰腺组织,以及阿尔茨海默症小鼠模型进行了大规模调节网络分析。最终,利用网络分析结果与标准分析(如聚类和差异表达分析)相结合,发现了许多健康和疾病系统的关键调控因子。本研究使得单细胞调控网络分析首次完整、有效、高通量和以疾病为中心的得以应用,极大地丰富了从scRNA-seq技术中获取更多有价值的内容。
实验结果:
与传统bulk转录组测序结果不同,单细胞数据固有的噪音和高度稀疏性,阻碍了基因间相关性可靠度量标准系数,例如Pearson、Spearman或者Cosine相关性分析的有效使用。因此,本研究开发了一种基于计算框架的新的相关性测量方法,专门用于单细胞数据分析,其基本原理是两个相关基因遵循细胞亚型之间相似的差异表达模式。因此,该方法不使用原始变量,即标准化的表达量计数(Expression Counts)来寻找关系,而是通过将表达量计数用Z-scores代替,来计算转换变量(Transformed Variable)之间的相关性(图1)。
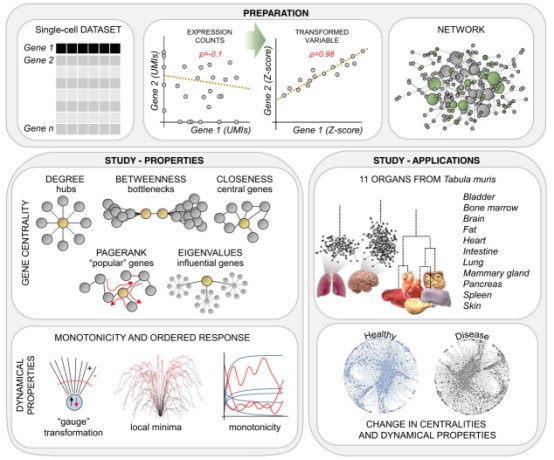
图1、计算框架方法概况图
当应用于由7697个小胶质细胞组成的数据集时,最终确定了933936个显著的基因间相关性(Pearson > 0.8),与标准化UMI计数数据相比,增加了近40000倍(图2,只有24个相关性)。
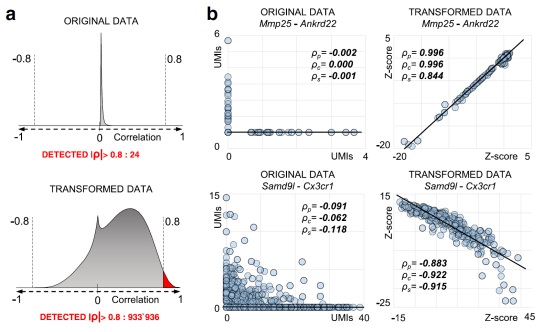
图2、利用该度量方法可以检测到单细胞数据中隐藏的相关性
在确定基因对基因的相关性后,应用适应性阈值法来保留显著相关性,并结合GO功能注释来推断获得相关调控网络。随后,研究者对计算框架方法得到的相关性结果进行了特异性、敏感性、假阳性等检测,结果都比较理想。
最后,为了评估使用从单细胞推断的大规模调控网络来帮助对scRNA-seq数据集进行生物学解释的价值,研究者首先将本方法应用于分辨小鼠器官的单细胞图谱。研究从11个器官产生调节网络:内胚层(肺、胰腺、肠)、中胚层(心脏、脂肪、脾脏、膀胱、骨髓)和外胚层(皮肤、大脑、乳腺)。最终结果正如预期的那样,网络中的节点数量(即基因)与检测到的基因的平均数量成比例。所有的网络都具有模块化特征,模块化程度最高的网络是胰腺、心脏和大脑,骨髓模块化程度最低。有趣的是,网络的密度(边/节点比率)变化很大。较低的网络密度可能表明频繁使用“间接”转录调节,即涉及没有直接基因调节功能的基因的信号级联。值得注意的是,网络密度和模块性呈反比关系,表明稀疏网络(如大脑、心脏和胰腺)以模块间连通性降低为代价,保持了强大的模块内连通性。因此,模块化一定程度上代表了组织异质性,表型多样性的增加与模块化的网络增多有关。
研究者推测调节网络对于潜在疾病相关的调节变化将是特别有用的,这些变化在当前的分析方法中是无法实现的。因此,研究者为来自糖尿病患者的2491个单细胞转录组建立了健康和T2D调节网络,最终研究发现,利用该方法可以检测到2型糖尿病(T2D)患者胰腺调节网络结构的改变(图3)。
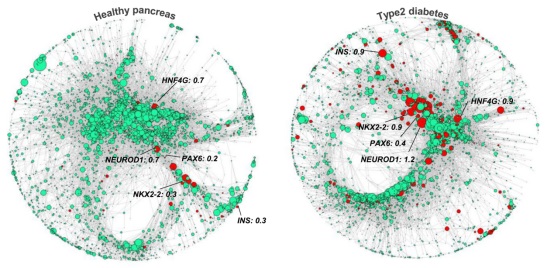
图3、2型糖尿病(T2D)胰腺单细胞数据的基因调控网络变化图
最后,研究者进一步评估了基于网络的方法在不同疾病和物种环境中的适用性,对来源于5XFAD转基因小鼠(阿尔茨海默症常用小鼠模型)的免疫细胞(CD45+)的scRNA-seq数据进行了分析。该数据集包含来自不同疾病阶段(1–8 months、对照和5XFAD)的22951个单细胞的转录组数据,通过对调控网路的观察,发现AD中的连通性普遍丧失,这增加了网络稀疏性和信号传播时间(最短路径)。因此,与对照相比,几个基因在阿尔茨海默病网络中的中心性不同。
研究结论:
1、提出了一个从单细胞转录组数据推断大规模基因调控网络的分析框架。
2、使用图论中的工具研究了网络的全局和局部性质,实现了全面的结果表征。
3、在健康和患病样本的单细胞数据集中生成了大量多样的调控网络结果,验证了这种方法的可行性。
关于天昊
天昊生物具有多年基因组、转录组和表观组检测与分析经验,现推出的10x单细胞转录组测序可为您提供专业便捷的科研服务及个性化的单细胞信息挖掘,期待成为您单细胞测序分析的优质服务提供商!
欢迎联系我们具体咨询!
邮箱:techsupport@geneskies.com
电话:400-065-6886