近日,福建医科大学附属第一医院神经内科陈万金教授和王柠教授团队最新研究汇报了常染色体显性遗传性痉挛性截瘫(HSP)的新亚型及其新致病基因UBAP1,这一成果于6月15日在线发表于国际神经病学与神经科学权威杂志《Brain》(IF=10.848)。基于对闽南地区112个家系样本的收集和基因测序,研究团队最终确定了HSP的新致病基因UBAP1,并基于斑马鱼模型,小鼠条件敲除,进一步明确了该致病基因的生物学功能。天昊生物有幸在该项研究中,参与了部分样本的全外显子测序和部分数据分析工作。
中文题目:UBAP1基因无义突变可导致单纯性常染色体显性遗传性痉挛性截瘫(HSP)
英文题目:Stop-gain mutations in UBAP1 cause pure autosomal-dominant spastic paraplegia
研究背景
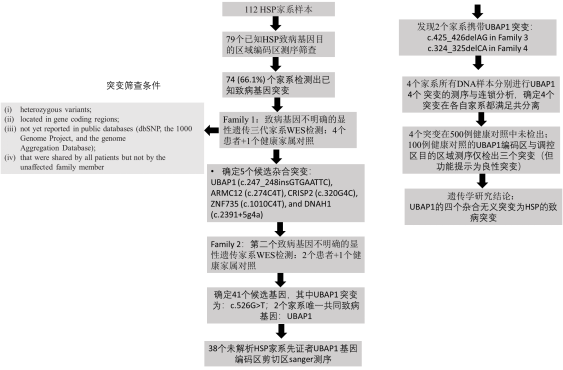
图1:HSP显性家系致病突变的遗传学筛查方法
研究结果
(1)遗传学分析,确定4个无关HSP显性家系的致病位点均为UBAP1 基因4号外显子的无义突变
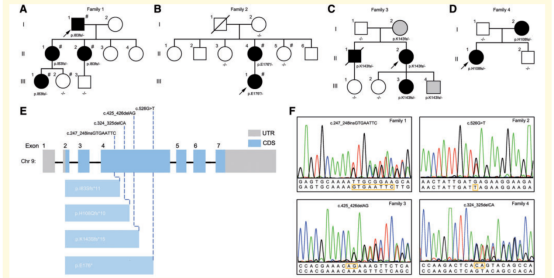
图2:4个HSP显性家系的4个UBAP1 基因无义突变(#标注为进行WES检测的患者)
(2)UBAP1 基因突变HSP患者的临床特征
(3)ubap1敲减斑马鱼模型,表现出明显的体型异常、胚胎发育异常、运动神经元生长受限、运动能力减弱以及寿命缩短。
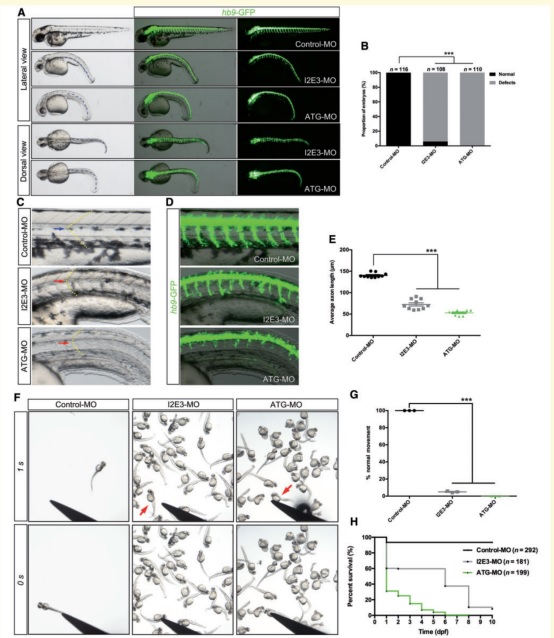
图3:ubap1敲减斑马鱼模型,表现出明显的体型异常和胚胎发育异常(ABCD)、运动神经元生长受限(E)、运动能力减弱(FG)以及寿命缩短(H)
(4)截断型UBAP1蛋白导致内体泛素化过程受损
ESCRT是一种能够识别并分拣泛素化蛋白质货物的蛋白复合体, ESCRT-I为其中一个亚复合体。而UBAP1编码的蛋白即为ESCRT-I的一个亚基,参与泛化物结合和内体分拣过程。但是基于突变型蛋白CO-IP研究发现,截断型UBAP1蛋白并未影响UBAP1与ESCRT-I其它亚基的结合。进一步在Hela细胞系中的研究发现,截断型UBAP1蛋白实际上破坏了细胞内早期内体处理和/或分拣泛素化蛋白质的过程。
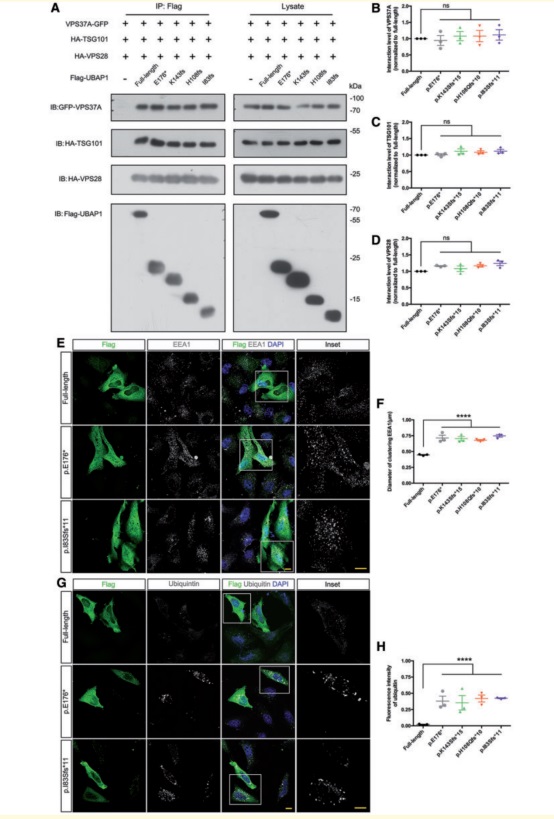
图4:截断型UBAP1蛋白并未影响UBAP1与ESCRT-I其它亚基的结合(ABCD),但破坏了细胞内早期内体处理和/或分拣泛素化蛋白质的过程(EFGH)。
(5)构建Ubap1flox/flox小鼠,对小鼠原代皮层神经元进行Ubap1条件敲除,损伤神经元胞体及轴突出现明显增大的内体以及异常蓄积的泛素化蛋白,最终加速了皮层神经元的凋亡。而体外在敲降小鼠皮层神经元中过表达人源正常UBAP1蛋白,则可以逆转部分内体相关转运障碍表型。
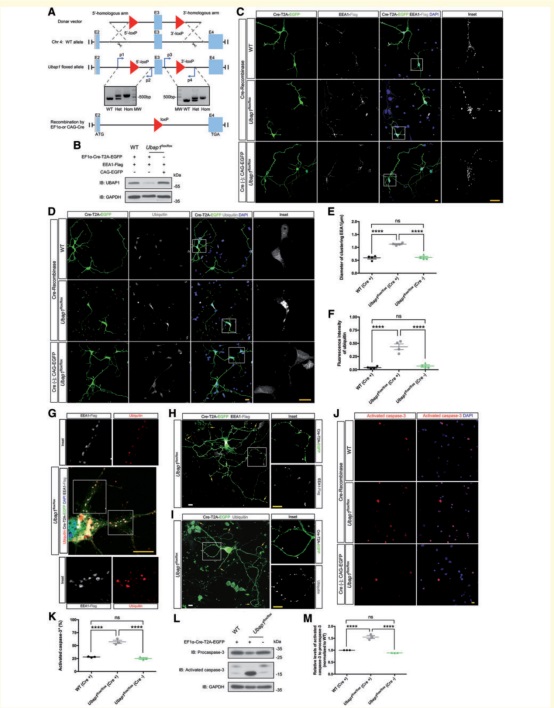
图5:小鼠原代皮层神经元进行Ubap1条件敲除,损伤神经元胞体及轴突出现明显增大的内体以及异常蓄积的泛素化蛋白(EFGHI),最终加速了皮层神经元的凋亡(JK)。
研究结论与意义