南京农业大学近期在土壤学领域一区杂志《Biology and Fertility of Soils》上发表了关于8年施氮对小麦根际细菌群落遗传效应的研究文章。在这项研究中天昊生物有幸承担了样品的相对定量扩增子测序工作。在恭喜客户发表文章同时,我们想跟大家分享一下文章的研究思路。
英文题目:Legacy effects of 8-year nitrogen inputs on bacterial assemblage in wheat rhizosphere中文题目:8年施氮对小麦根际细菌群落的遗传效应期刊名:Biology and Fertility of Soils影响因子:4.829
研究概要
本研究主要探讨8年施氮(以0、140、280、470和660kg N ha−1 year−1为梯度)对小麦根际细菌群落多样性、相互作用和组装过程的影响。根际细菌α-多样性随历史施氮量的增加而增加,但在施氮量超过280kg N ha−1 year−1时,根际细菌α-多样性没有变化。历史施氮量明显改变了根际细菌群落组成,施氮量大的土壤与不施氮的土壤差异较大。最接近相对指数(NRI)和最接近分类单元指数(NTI)分析表明,大多数样品的根际细菌群落是系统聚类的,高氮处理(>470 kg N ha−1 year−1)比低氮处理(<140 kg N ha−1 year−1)表现出更高的聚类水平,表明历史氮输入越高,土壤环境选择压力越大。小麦地上生物量的增加伴随着共生网络规模和连接性的增大。总体而言,随着历史氮输入的增加,所产生的遗留效应迫使根际细菌群落承受更高的环境选择压力,并在随后的作物生长过程中间接影响小麦根际组合的复杂性。
研究背景
氮(N)肥是限制植物和微生物生长的重要营养物质之一,在全球范围内被供应到陆地生态系统中,自工业革命以来,由于人类活动的结果,氮的输入率翻了一番。在我国集约农业地区,相当数量的氮(550-600 kg N ha−1 year−1)直接施入农田土壤。氮素的富集会影响土壤微生物群落的组成和功能,随着群落组成的变化,微生物多样性和呼吸作用降低,主要受土壤pH、有机碳和总氮含量的变化驱动。除了对土壤微生物的影响外,施氮还增加了与植物相关的凋落物C和根际沉积物C。由于长期施用肥料而引起的土壤变化称为遗留物或残留物,即使停止施肥,也可能在随后的季节产生持续效应(称为遗留效应)。先前的研究表明,先前的植物作物类型和土地利用历史对微生物群落组成和多样性具有遗留影响,然而,目前还缺乏对长期施氮对微生物多样性影响的研究。根际是植物根系周围的毫米级区域,是绝大多数活性微生物的栖息地,因此也是微生物相互作用的热点。根际微生物以根系分泌物为食,被认为是植物表型的一部分,在功能生态学中起着关键作用过程,如获取植物养分,凋落物分解,保护土壤健康。研究发现植物的种类、基因型和发育阶段可以改变根际微生物的群落组成和多样性,从而影响土壤功能。此外,土壤盐分和pH值等理化性质的变化也会对根际微生物群落产生影响。因此,了解根际细菌群落对长期施氮遗留效应的响应是非常重要的。
以前在研究根际群落时,主要采用了基于分类单元和功能的方法,然而,细菌系统发育可能提供更多关于基于性状的群落组装的信息,群落组装过程包括确定性过程(例如,系统发育聚类和系统发育过度扩散)和随机过程(例如,概率扩散和生态漂移)。环境过滤会导致物种的亲缘关系比预期的更为密切(系统发育聚类),而竞争性排斥则会导致亲缘关系不太密切的物种(系统发育过度分散)。一些研究表明,在细菌群落中,确定性过程占主导地位,随着海拔、盐度梯度、极端酸性或碱性条件的增加,系统发育聚类增加,观察到的群落聚集模式也取决于它们是在大空间尺度上进行检查还是在精细空间尺度上进行检查。目前,韦伯等人提出了基于零模型的群落组合系统发育结构分析方法,即观察到的平均成对系统发育距离(MPD)和平均最近分类单元距离(MNTD)分别与零模型期望值进行比较,这种方法为识别群落组装过程提供了一种很有前途的选择。
微生物群落组装的生态过程不仅包括环境因素引起的系统发育群落结构的变化,还包括微生物间的相互作用。这些关系可以包括积极的(如互惠)、消极的(如竞争)和中立的关系。微生物之间的相互作用对土壤养分循环和作物健康具有潜在的影响。共生网络分析为探索微生物间相互作用对环境的响应提供了有力的工具。同时,网络分析可以用来识别支持微生物群落功能和稳定性的关键物种;例如,在底土中,一些关键属的消失可能导致群落破碎。因此,通过共现网络分析研究微生物的相互作用,有助于了解不同环境下群落聚集的基本原理,识别群落中可能存在的关键类群。
土壤取样与试验设计:
在中期定位试验场,以冬小麦、夏水稻的轮作为主,采集了不同施氮量的土壤,从2010年开始实施现场试验。试验采用随机区组设计:3个重复,5个氮肥处理,历史施氮8年,每年施氮量分别为:0、140、280、470、660 kg N ha−1(以下分别为N0、N140、N280、N470、N660),栽培冬小麦和夏水稻。在整个试验过程中,尿素供氮,过磷酸钙供磷,氯化钾供钾。2017年小麦开花期,采用固体细沟采样器(内径3cm)和随机取样方法,从每个处理区0-20cm土层中采集土壤5kg,风干土样用不锈钢网(<5mm)过筛,每种氮处理的土样分别用聚乙烯袋充分混合。盆栽试验前测定了各处理的土壤理化性质。
将土壤填入聚丙烯盆中(高度×直径,10 cm×12 cm),土壤约500 g干重,进行了16个重复的微观实验,以尽量减少重复之间的变异性,本研究共安排80盆(5次施氮量×16次重复)。将盆栽置于玻璃温室中,每天12小时,平均气温25°C,在盆上播种小麦,每天用自来水浇水,保持土壤水分在20%左右。为了消除肥料对土壤微生物和植物的干扰,研究历史氮素输入对土壤微生物和植物的遗传效应,在整个植物生长期不施肥。
16S扩增子相对定量测序:
在开花期(移栽后8周生长),收集80个盆栽植物,对根际土壤进行采样。根际土壤样品的采集方法是将包括根在内的整个小麦植株从土壤中移开,通过抱枝的方式进行剧烈摇动,并将残留在根表面的土壤视为根际土壤。包括根际土壤的根系立即从植株上切下,转移到含有10毫升无菌水的无菌离心管中。附着在根表面的土壤通过涡旋和振荡混合到水中,然后除去根,将装有悬浮液的管子涡旋5分钟,11000g离心约5 min,弃上清液,剩余根际土壤部分保存于-20℃,48 h内提取DNA。使用MoBio Powersoil DNA Isolation Kits提取根际土壤DNA,然后进行16S扩增子相对定量测序(V4-V5区域)。
植物和土壤性质检测:
将地上植物和根样品在75℃下烘干,直到获得用于生物量测量的恒定重量。土壤样品经风干和筛分进行化学分析,例如测定土壤pH值、有效磷(AP)、有效钾(AK)、有机碳(SOC),总氮(TN)、碱解氮(Alk-N)等指标。
研究结果
植物生物量与土壤特性
盆栽试验前,N0处理与其它施氮处理的土壤化学性质存在显著差异。N0处理土壤AP、AK含量及pH值均显著高于其它4个施氮处理。长期施氮与不施氮相比,提高了土壤NO3-–N和TN含量。盆栽试验结束时,各处理间土壤理化性质的差异保持不变(表1)。与其它氮肥处理相比,N0处理的土壤化学性质和植物生物量均表现出显著差异(表1)。土壤pH值在N0时表现出最大值(pH=7.47),N660为最低值(pH=7.25)。四种施氮处理的土壤有机碳SOC和全氮TN含量均高于N0处理。长期施氮增加了土壤Alk-N浓度(从102.13到122.04),显著降低了土壤有效磷AP(从57.14到38.33)和有效钾AK(从90.98至64.17)。施氮处理植株总生物量显著高于N0处理,但施氮处理间差异不显著。N140、N470和N660处理的地上生物量显著高于N0处理。虽然不同处理间的根系生物量存在显著差异,但没有观察到一致的趋势。
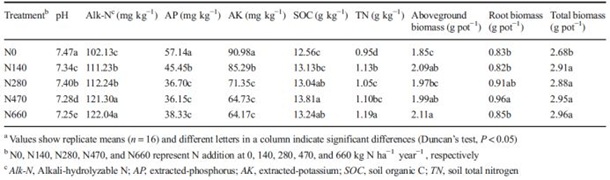
表1种植末期各处理土壤理化性质及植物生物量
遗传效应驱动小麦根际细菌群落的多样性和差异性
根际细菌Chao1、Shannon和观测丰富度指数与历史施氮量显著相关(图1)。低氮处理(N0和N140)的样品多样性显著低于高氮处理(N280、N470和N660)。根据拟合模型,Chao1指数、Shannon多样性和观测丰富度随施氮量的增加呈非线性增加(图1)。
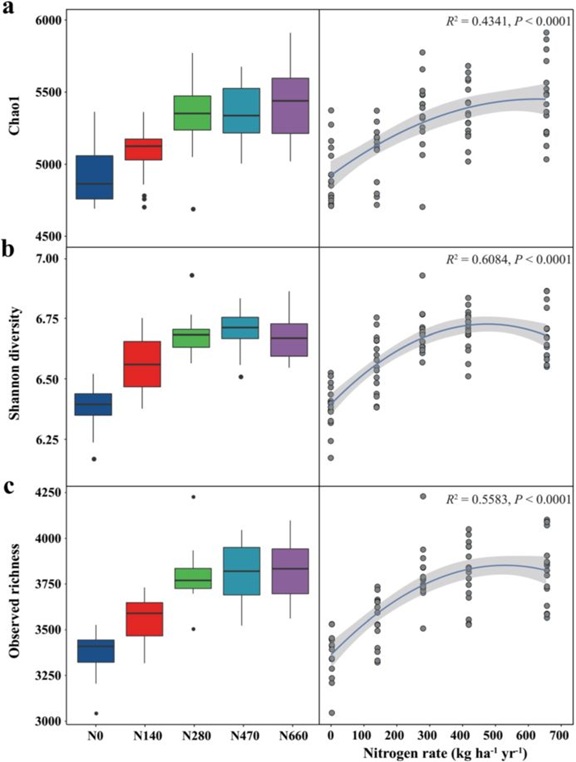
图1根际土壤细菌α-多样性在N输入梯度上的变化,包括Chao1指数(a)、Shannon多样性(b)和观测丰富度(c)。
为了评估根际细菌群落的差异(β-多样性),使用基于Bray-Curtis和加权UniFrac距离的度量来评估群落组成。NMDS图显示根际细菌群落在不同施氮量下有明显的聚集性(图2)。同时,MRPP、ANOSIM和ADONIS试验表明,所有处理之间的群落组成存在差异。所有样本的差异配对比较表明,随着氮输入的绝对差异,细菌群落组成(Bray-Curtis距离)和系统发育(加权UniFrac)距离的差异显著增加,然而,组内差异没有显著变化。
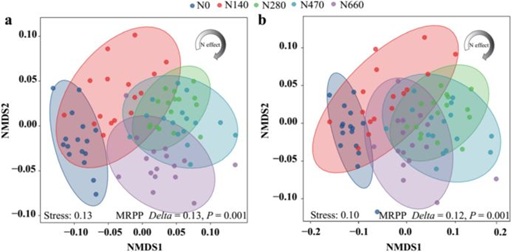
图2 基于Bray-Curtis距离(a)和加权均匀度(b)的NMDS分析显示处理间土壤根际细菌群落组成的变化。基于细菌距离矩阵的组间MRPP差异检测。
遗传效应驱动小麦根际细菌群落组成与功能
为了评价根际细菌群落组成,对30个门进行了高质量序列分析,属于15个最丰富门(相对丰度>1%)的细菌占所有序列的83.8%,其中变形菌门占从每个样本获得的细菌总序列的29.5-48.1%。基于相对丰度和观测到的OTU的数量,在所有土壤样品中检测到优势细菌菌群(变形杆菌、拟杆菌、嗜酸细菌、氯磷菌、厚壁菌和放线菌),但它们的相对丰度变化显著。然而,在门的水平上,随着氮的添加,观察到两种不同的相对丰度模式,随着氮输入速率的增加,Deltaproteobacteria的相对丰度不断增加,而Verrucomicrobia的相对丰度则相反。在低氮条件下(小于280 kg N ha−1 year−1),某些菌的相对丰度显著降低或增加,但当氮沉积速率超过280kg N ha−1 year−1时,这些菌则没有变化,例如Gammaproteobacteria, Bacteroidetes, Acidobacteria, Chloroflexi, Gemmatimonadetes,其它细菌在氮梯度上没有显示出一致的趋势。此外,还确定了不同处理间显著差异的优势属(相对丰度>1%)。然而,氮处理下优势属的变化并不总是在门水平上表现出来,例如,属于Alphaproteobacteria亚门的Rhodobacter和属于Firmicutes的Bacillus的变化并没有分别导致Alphaproteobacteria和Firmicutes的一致变化。历史高氮增加了Gemmatimonas, Gp4, Gp6, Gp7的比例,而降低了Ohtaekwangia, Rhodobacter, Pseudoxanthomonas的比例。
FAPROTAX工具用于将细菌分支(如属或种)映射到代谢和生态相关功能,本研究中的细菌可被分配到66个功能群。在预测的功能组中,33个与C、N和S循环相关的主要功能组被历史氮输入显著改变。历史高氮输入与低氮相比,遗留效应导致了与氢营养甲烷化、甲烷化、好氧氨氧化、好氧亚硝酸盐氧化、硝化作用、硫代硫酸盐呼吸和暗硫化物氧化相关的功能群的相对丰度较高。在高氮处理(N280、N470和N660)中,与纤维素分解、甲醇氧化、硝酸盐还原有关的功能群的相对丰度均显著低于N0和N140处理。此外,N0处理中甲壳分解功能群的相对丰度最大。
氮梯度下根际细菌群落的NRI和NTI
根据零模型分析,与高氮处理(超过280 kg N ha−1 year−1)相关的最接近相对指数(NRI)和最接近分类单元指数(NTI)显著大于零(图3)。这表明高氮施用的遗留效应使物种间的亲缘关系比预期的更为密切。NTI值显著大于零与低氮处理(小于280 280 kg N ha−1 year−1)相关(图3b),而低氮样品中细菌群落的NRI值与零无显著差异(图3a)。这些结果表明,低氮处理根际细菌的系统发育聚类集中在系统发育树的末端,而群落组成的随机系统发育模式则发生在较高层次的树节点。
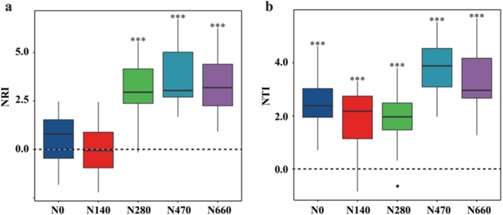
图3土壤中根际细菌群落的最接近相对指数(NRI)(a)和最接近分类单元指数(NTI)(b)随氮梯度的变化而变化。正的NRI和NTI值表明群落中的物种比预期的更接近。星号表示NRI(或NTI)值与显著性水平之间的显著差异
氮梯度上根际细菌群落共现网络
采用共生网络分析方法,研究了小麦根际细菌间潜在的相互作用和生态位共享。为了进行这一分析,我们基于5个施氮梯度土壤中的序列数据,在属(和OTU)水平的根际细菌分类群之间构建了5个细菌共生网络。共生网络图显示了不同处理间的巨大差异(图4a-e)。根际网络中基于属水平的节点数和连接数在N梯度上是可变的,最小值出现在N0处理中(即,节点数从N0的92到N660的193,连接数从N0的100到N140的1184)(表2,图4)。根际网络节点数随地上生物量的增加而单调增加(图4f)。与高地上生物量相关的根际网络比与低地上生物量相关的根际网络包含更多的连接,这表明高地上生物量产生更复杂的网络模式(连接数量,从100到1184)(图4a-f)。此外,在OUT水平,共现网络的节点和连接的变化与属水平网络的变化趋势相似。综上所述,随着土壤肥力的增加,根际细菌网络随着地上生物量的增加逐渐变得复杂。
此外,BC反映了一个特定节点对网络中其他节点交互作用的控制程度,BC得分高的属对维持微生物网络的连通性有很强的影响。与高地上生物量相关的根际网络中大多数节点的BC比与低地上生物量相关的根际网络中的BC强得多,并且随着地上生物量的增加,强节点的数量逐渐增加(图4f)。观察到的网络节点的BC分布表明,与高地上生物量(N140、N470和N660)相关的多个节点(例如:Caulobacter, Rhodobacter, Ignavibacterium, Nitrospira)承担了关键的网络作用,而一些节点(例如:Sporomusa, Clostridium_III, Bacillus, Tumebacillus, Geobacter)的BC值较低,与地上生物量(N0,N280)相关。细菌群落经过重新排列,在确定整个网络组成方面赋予了几个属以关键作用。在与高地上生物量相关的根际共生网络中,很大一部分节点定义了网络拓扑结构,表明更多的属形成了细菌群落相互作用的模式。
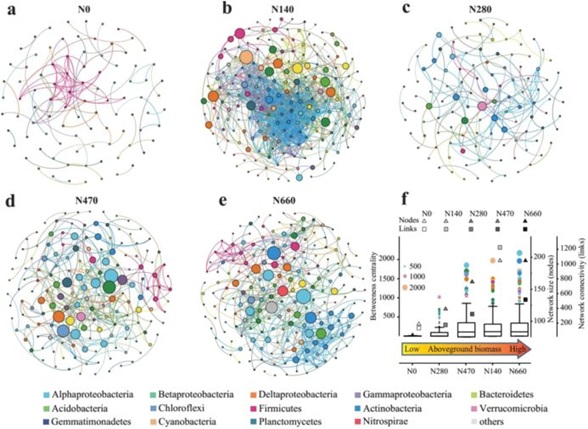
图4各处理根际细菌群落基于属水平的共现网络分析(a-e)。每个点代表一个细菌系统类型。每个节点的大小与中间中心度的个数成正比。节点间的中间中心度和网络复杂性(节点和连接的数量)随地上生物量(f)的变化。

相关链接:
喜讯!天昊生物微生物项目文章登陆《Environmental Pollution》;
喜讯!天昊微生物测序项目文章登陆Cell子刊《Current Biology》;喜讯!天昊生物16S扩增子绝对定量测序项目文章再次登陆《Science of the Total Environment》;
天昊微生物项目文章:【SBB】了解长期施加有机物料如何增加土壤磷酸酶活性:针对phoD和phoC功能性微生物种群的研究;